 I am currently an Assistant Professor in Computational Chemistry in the Department of Applied Sciences at Northumbria University.
I am currently an Assistant Professor in Computational Chemistry in the Department of Applied Sciences at Northumbria University.I obtained my PhD from the University of York in 2015 where I was subsequently appointed a Postdoctoral Research Associate with the groups of Dr. John Moore and Prof. John Goodby in 2015, working on combining experimental and computational studies of ordered systems in order to develop design methods to aid in identifying synthetic targets. I was appointed to my current role at Northumbria University in 2017.
Current and previous research
Sections below list largely academic research. Other industry-focussed research projects include collaborations with AkzoNobel and Palintest.
Rationalising reactivity of bioorthogonal reagents
In collaboration with Dr Valery Kozhevnikov
Contributors: Connor Atess, Numair Zaman, Aminah Shafiq
Bioorthogonal reactions are those that take place in-vivo without interfering with biological processes. Designing such reagents and understanding trends in reactivity can be aided by the use of ab-initio methods to calculate transition state energies, transition state conformations, frontier orbital energies etc.
Papers
Pyreno-1,2,4-triazines as multifunctional luminogenic click reagentsOrg. & Biomol. Chem., 2025, 23, 10127-10134

Dalton Trans., 2024, 53, 15501

Dalton Trans., 2023, 52 , 10927-10932

Chem. Commun., 2019, 55 , 14283-14286

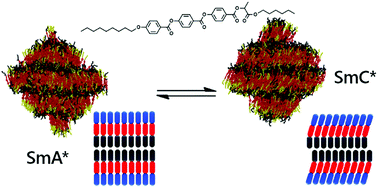
Modelling de Vries behaviour in liquid crystals
Contributors: Kristian Poll
Ferroelectric SmC* liquid crystals have the potential to be used widely in future display technologies, but the shrinkage they typically exhibit on tilting at the SmA/SmC transition is particularly problematic. A sub-class of these materials exhibit “de Vries” behaviour, in which no such shrinkage is observed, but the behaviour is poorly understood. We have used fully atomistic molecular dynamics simulations to gain an understanding of these intriguing materials.
Papers
Sub-layer rationale of anomalous layer-shrinkage from atomistic simulations of a fluorinated mesogenMater. Adv., 2022, 3, 1212-1223

J. Mater. Chem. C, 2020 ,8 , 13040-13052
Calculating ligand binding in lanthanide extraction
In collaboration with Dr Frank Lewis
Contributors: Anthony Sayer, Anya Nelson, Sam Taylor, Lauren Gardiner, Maddie Gilbey
Design of ligands that selectively bind actinides over lanthanides is a challenging problem. Use of ab-initio methods to calculate binding energies can aid sythetic chemists in understanding ligand behaviour and in designing new, improved materials.
Papers
Evaluation of Multidentate Ligands Derived from Ethyl 1,2,4-triazine-3-carboxylate Building Blocks as Potential An(III)-Selective Extractants for Nuclear ReprocessingChemistryOpen, 2025, 14 , e202400306

Chem. Eur. J., 2020, 26 , 428-437

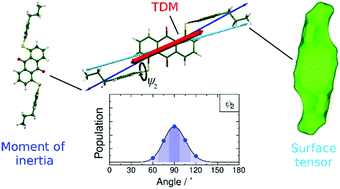
Elucidating structure-property relationships in dye-doped liquid crystals
Liquid-crystal dye mixtures have many potential applications including smart windows, privacy glass, and low-power displays. However, the understanding of dye behaviour is such mixtures and rational design of systems with desirable characteristics is an area that has received relatively little attention from a computational perspective. Combining classical methods with ab-initio approaches enables experimental alignment behaviour to be understood, and provides a route by which improved systems can be designed.
Papers
Dyes for guest–host liquid crystal applications: a general approach to the rapid computational assessment of useful molecular designsPhys. Chem. Chem. Phys., 2023, 25, 10367-10383

Liq. Cryst., 2017, 44, 2029-2045

Chem. Eur. J., 2017, 23 , 5090-5103

Phys. Chem. Chem. Phys., 2017, 19, 813-827
Liq. Cryst., 2016, 43, 2363-2374

Phys. Chem. Chem. Phys., 2016, 18 , 20651-20663

J. Phys. Chem. C, 2016, 120, 11151–11162

Chem. Eur. J., 2015, 21 , 10123-10130

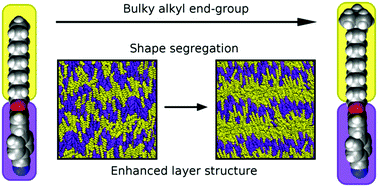
Quantifying and understanding self-organisation using x-ray scattering
The quantification of alignment in self-organising systems is often not straightforward. X-ray scattering theoretically provides significantly more information than many other methods, but data processing can lead to significant errors that some of our work has addressed. Subtle changes in molecular structure can heavily influence the molecular organisation in liquid crystals; a combinination of x-ray scattering experiments with classical simulations is a powerful tool with which such influences can be quantified and understood at a molecular and sub-molecular level.
Papers
Shape segregation in molecular organisation: a combined X-ray scattering and molecular dynamics study of smectic liquid crystalsSoft Matter, 2019, 15, 7722-7732
Liq. Cryst., 2017, 46, 11-24
